Industry view
China Entry for Medical Device and Life-Science Companies
NMPA medical device registration, clinical trial requirements, WFOE for direct sales vs distributor model. Realistic 18-30 month timelines.
Email Mike—
Y1 cost band
Mike's running benchmarks
—
Time to first revenue
Mike's running benchmarks
—
Common pivot at month 12
Mike's running benchmarks
</p>
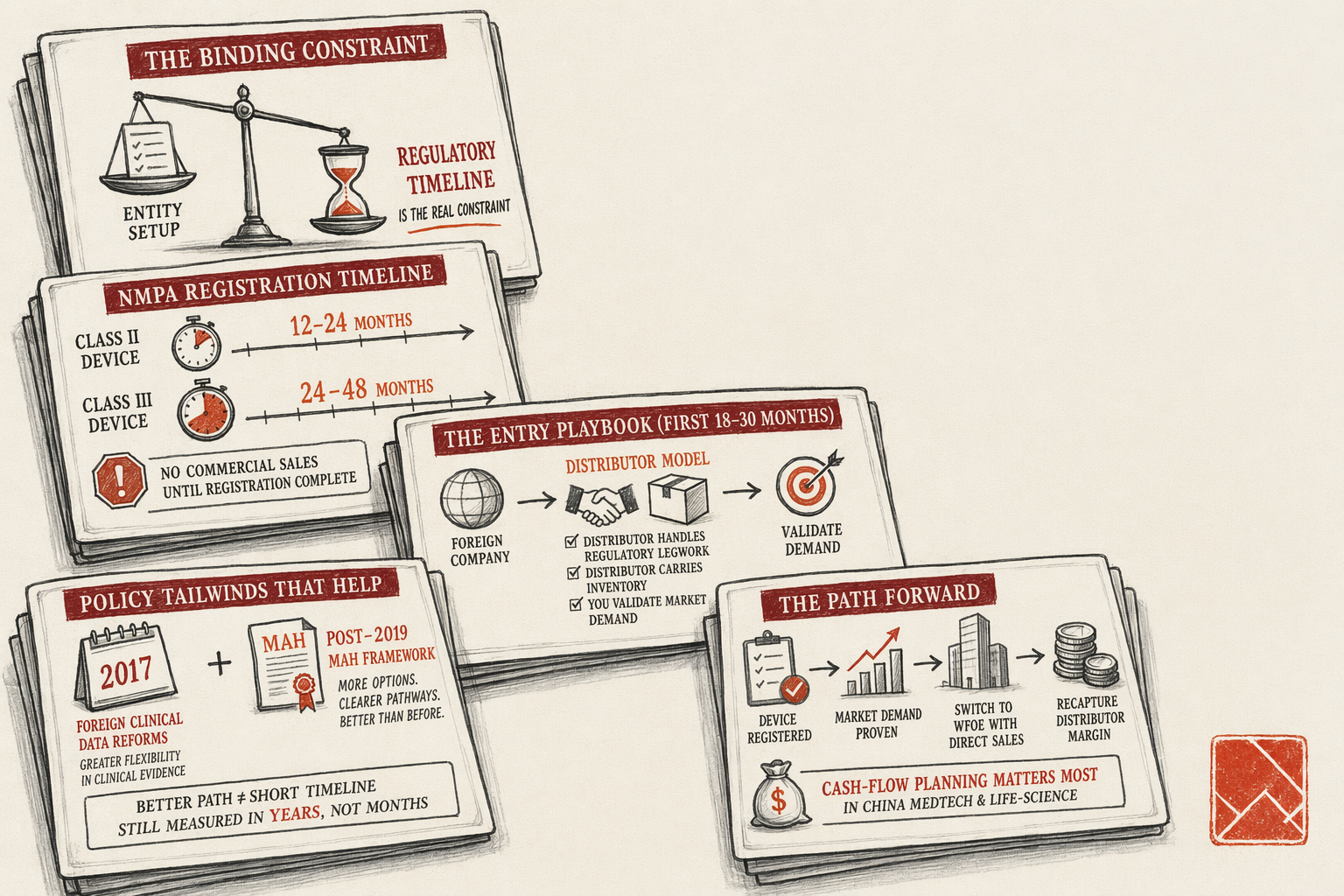
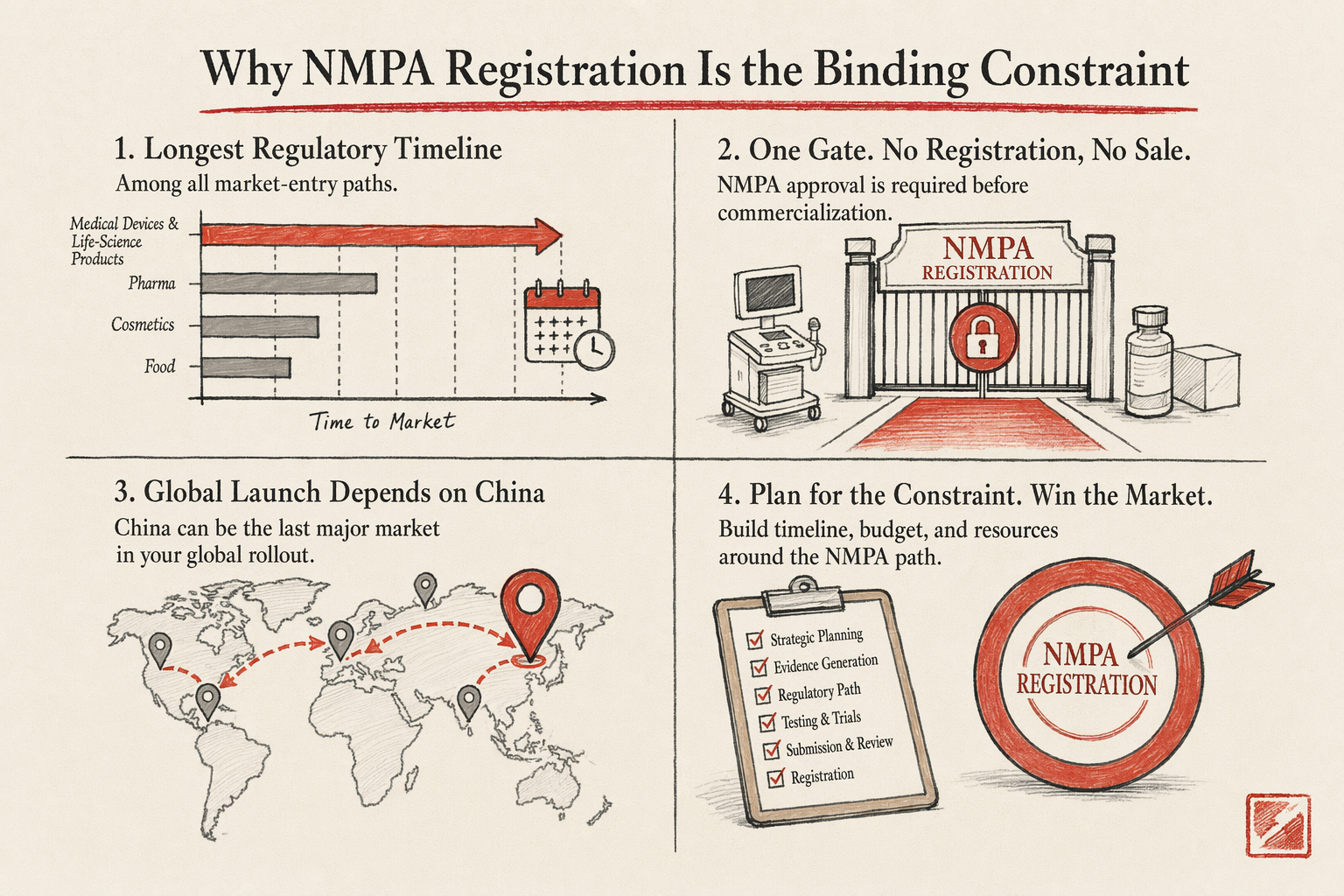
Medical devices and life-science products face the longest regulatory timeline of any foreign-brand market-entry path into mainland China. NMPA registration is the binding constraint on the project — you do not sell commercially until the device or product is registered with NMPA (National Medical Products Administration, the mainland regulatory authority that absorbed the former CFDA functions in 2018). For Class II devices the registration timeline is typically 12-24 months from a clean submission. For Class III devices it is 24-48 months, with clinical-trial requirements that can compound further. Foreign founders entering this market need to plan on multi-year horizons, with cash-flow planning that reflects the regulatory wait.
The compensating factor is that the foreign-entrant path has materially improved since the 2017 reforms accepting foreign clinical-trial data and the 2019 marketing-authorization-holder (MAH) framework. What was a near-impossible market for first-time foreign medical-device entrants in 2010-2015 is now a navigable (if still slow) regulated market for well-prepared sponsors. This page walks through what medical-device and life-science companies actually need, the NMPA regulatory layer in operational detail, the structural pattern that fits most foreign entrants, the rejection-and-stall patterns we see, and the realistic timeline and budget for a sequenced engagement.
What medical device and life-science companies typically need
The operational ask from a foreign medical-device or life-science company entering mainland China tends to cluster into five categories.
One — NMPA registration of the device or product. The single longest and most resource-intensive workstream. Registration documentation includes product technical files, manufacturing-process documentation, clinical-trial data (foreign and possibly mainland), safety and biocompatibility testing reports, labeling and instructions-for-use translated and adapted for mainland market, post-market-surveillance plan, and quality-management-system certifications (ISO 13485 for devices, and mainland-specific GMP equivalents). The registration is filed with NMPA's relevant device-evaluation center; review periods run 12-48 months depending on class and complexity. Without NMPA registration, the device cannot be legally marketed or sold in mainland China.
Two — a registration holder entity. Under the marketing-authorization-holder framework that mainland China implemented starting 2019, the registration can be held by an entity separate from the manufacturer. For foreign companies, the most common pattern is to hold the registration in a mainland-based entity (WFOE or in the early stages a distributor that holds the registration on a contractual basis) while the manufacturer remains the overseas parent. The registration holder bears regulatory obligations including post-market surveillance, adverse-event reporting, recall coordination if applicable, and the legal-representative-of-record role for NMPA communications. The choice of registration holder is strategic — distributor-held registrations create dependency, WFOE-held registrations create operational commitment.
Three — a clinical-data strategy. Class II devices that are well-characterized and have foreign clinical-trial data following ICH-GCP standards can typically register on the foreign data alone under the 2017 reforms. Class III devices may still require confirmatory mainland clinical trials, though the threshold has dropped substantially. The clinical-data strategy decision — register on foreign data alone, register with a small mainland confirmatory trial, or commission a full mainland trial — depends on the device class, the strength of the foreign data, and the specific NMPA pre-submission guidance for the device category. Engage an NMPA-experienced regulatory consultant during the pre-submission phase to shape the clinical-data strategy correctly; this saves more time later than any other single project decision.
Four — a commercial structure for sales when registration completes. The distributor model is the typical first commercial structure: a mainland medical-device distributor (often a specialist in your device category) handles regulatory filings, inventory commitment, sales-team coverage, post-sales support, and the hospital-and-clinic-channel relationships that medical-device commerce runs on. Distributor margin ranges from 25-50% depending on category and exclusivity terms. Once the device is selling consistently and the market demand is validated, the WFOE-with-direct-sales conversation activates. The WFOE typically transitions in 18-30 months from initial commercial sales, with the distributor relationship evolving to a sub-distributor or transitioning out entirely.
Five — quality-management-system certifications recognized by NMPA. ISO 13485 (the medical-device-specific quality management standard) is the international baseline. Mainland Chinese regulatory practice recognizes ISO 13485 as part of the technical-file submission but additionally requires evidence of manufacturing-quality controls that may need mainland-specific inspections (NMPA quality-system inspections, or in some cases on-site inspections at the foreign manufacturing facility). Plan for the quality-system inspection workstream as a parallel track to the registration workstream — the inspections typically occur during the registration review period and missing the inspection-readiness window can delay registration by 6-12 months.
The vertical's specific regulatory layer — NMPA Class I/II/III, clinical trial data, GHTF reform
Beyond the generic entity work, medical-device and life-science companies face the most demanding regulatory layer of any foreign-brand vertical. Four regulatory regimes define the path.
NMPA device classification (Class I, II, III). Class I devices are low-risk and follow a notification-based filing (备案), typically completing in 3-6 months. Class II devices are moderate-risk and follow a registration (注册) process with clinical-data requirements, completing in 12-24 months. Class III devices are high-risk (typically including implantables, life-support equipment, novel diagnostics, and devices with significant safety considerations) and follow the most rigorous registration process, completing in 24-48 months with often-mandatory mainland clinical trials. The classification of a specific device follows the NMPA Classification Catalog (医疗器械分类目录), which is updated periodically. Mis-classification at the start of the project — declaring a Class III device as Class II to chase the shorter timeline — fails at the technical review stage and forces a restart. Determine classification accurately during diligence.
Clinical-trial data acceptance. The 2017 Notice on the Acceptance of Foreign Clinical Trial Data (临床试验数据接受公告) substantively reformed the foreign-data acceptance regime. Foreign clinical-trial data that follows ICH-GCP standards, that is sufficient to demonstrate safety and efficacy for the mainland intended-use population, and that includes Chinese-population representation either through ethnic Chinese subjects in the original trial or through supplementary mainland data, can be accepted as the primary clinical-data basis for NMPA registration. This applies most clearly to Class II devices and to Class III devices where the device is well-characterized and the foreign data is robust. For Class III devices in novel categories or with limited foreign data, mainland confirmatory trials may still be required. The pre-submission consultation with NMPA's relevant device-evaluation center is the diagnostic for which path applies to your specific device.
GHTF (Global Harmonization Task Force) alignment. Mainland Chinese medical-device regulation has progressively aligned with GHTF principles since the mid-2010s. The classification system mirrors GHTF risk-based classification. The quality-management-system requirements align with ISO 13485 international standards. The post-market surveillance requirements align with international medical-device-vigilance practices. The cumulative effect for foreign entrants is that documentation, processes, and quality controls that meet US FDA or EU CE-mark requirements are increasingly transferable to NMPA submissions with translation and mainland-specific adaptation, rather than requiring de-novo mainland-specific development.
Marketing-authorization-holder (MAH) framework. Implemented for pharmaceuticals starting 2019 and progressively extended to medical devices, the MAH framework permits the registration holder to be a separate entity from the manufacturer. For foreign companies, the MAH framework allows a mainland-based WFOE or registration holder to hold the device registration while the manufacturing operations remain overseas. The MAH bears the regulatory and post-market-surveillance obligations, which separates the regulatory exposure cleanly from manufacturing operations. The MAH is a meaningful improvement on the pre-2019 system that required the manufacturer to be the registration holder, which forced foreign manufacturers to either set up mainland manufacturing or to rely on distributor-held registrations with attendant strategic dependency.
Typical entity-structure pick — distributor model first, WFOE on direct-sales readiness
The default structure for foreign medical-device and life-science company mainland market entry runs in two stages.
Stage one — distributor model for the first commercial phase (months 0-24). The first commercial structure for most foreign medical-device companies is a distributor relationship rather than a WFOE. The structural rationale: NMPA registration is the binding-path workstream and runs 12-48 months independent of entity choice. During the registration period, the foreign company benefits from having a mainland-based partner who can handle the regulatory paperwork, the mainland-side communications with NMPA, the eventual distributor-channel sales-team coverage, and the inventory commitment that direct-sales-WFOE would otherwise carry. The distributor model also lets the foreign company validate market demand before committing operating exposure.
Distributor selection is strategic. Categories of distributors: large national medical-device distributors (operating across multiple device categories and regions), specialist distributors (concentrating on a specific device category with deep channel relationships in that category), regional distributors (geographic specialists with strong hospital and clinic networks in their region). Selection criteria include category specialization, hospital-and-clinic channel reach, registration-handling experience, exclusivity terms preferred, and margin model. Distributor margin typically runs 25-50% of selling price depending on category and exclusivity. Distributor-held registrations create strategic dependency — exclusivity-period clauses, transfer-of-registration clauses, and exit-rights provisions in the distributor agreement are the key contract-negotiation points.
Stage two — WFOE conversion for direct sales (months 18-36 onwards). The WFOE conversion activates when several conditions converge: the device is NMPA-registered, mainland market demand is validated through distributor-channel commercial sales, the foreign company has a strategic decision to commit operating presence to the mainland market, and the distributor margin is large enough that recapturing it through a WFOE direct-sales operation justifies the operational overhead. The WFOE setup mirrors the trading-WFOE pattern from the industrial-manufacturers vertical: registered in an appropriate jurisdiction (Shanghai, Beijing, Shenzhen, Guangzhou), general-taxpayer VAT status, customs registration if importing finished devices, import-export operating right, SAFE FX registration, mainland sales-team build-out.
The transition from distributor to WFOE-direct-sales is the strategically delicate phase of the engagement. Done abruptly, it destroys the distributor relationship and risks losing the hospital-and-clinic channels the distributor built. Done thoughtfully, the distributor evolves to a sub-distributor handling specific regional or channel segments while the WFOE captures the higher-margin direct-sales channels. The transition typically runs 6-12 months with both structures operating in parallel before the distributor's exclusivity period ends or is renegotiated. Engage WFOE registration and the regulatory transfer-of-registration workstream simultaneously, because transferring the NMPA registration from the distributor's name to the WFOE is a multi-month administrative process at NMPA.
Common rejection patterns specific to medical devices and life sciences
The rejection and stall patterns specific to medical-device and life-science mainland entry cluster around four themes.
Pattern one — clinical-data inadequacy at the NMPA technical review. The foreign clinical-trial data submitted is judged insufficient by NMPA's technical reviewers — either because the data does not include sufficient Chinese-population representation, because the trial methodology does not meet current NMPA expectations, or because the device's intended-use claims in the mainland market diverge from the foreign labeling. The fix is to engage NMPA pre-submission consultation early (typically 6-12 months before the formal submission) to map the clinical-data strategy to NMPA's current expectations for the device category, and to commission supplementary mainland data or expanded foreign data analysis if gaps are identified pre-submission. Reactive supplementation after a rejection adds 12-24 months to the registration timeline; proactive consultation adds 3-6 months and substantively de-risks the submission.
Pattern two — quality-system inspection failure during the registration review. NMPA quality-system inspections during the registration review identify gaps in the manufacturer's quality-management system — typically gaps in the mainland-specific documentation, in the post-market-surveillance plan readiness, or in the on-site quality-control processes. The fix is to prepare the quality-system inspection package with mainland-specific requirements in view from the start, including translated SOPs, mainland-adapted post-market-surveillance plan, and inspection-readiness rehearsals if applicable. Many foreign manufacturers underestimate the mainland-specific quality-documentation overlay on top of their existing ISO 13485 compliance.
Pattern three — strategic dependency from distributor-held registration. The distributor holds the NMPA registration in their entity's name, and at the end of the initial commercial period the distributor leverages the registration ownership to extract more favorable exclusivity terms, to block the foreign company from transitioning to direct sales, or to negotiate a release-payment for transferring the registration. The fix is the distributor-agreement structure at the start of the engagement: registration-ownership clauses, transfer-of-registration provisions on defined triggers, exit-rights clauses that preserve the foreign company's optionality. Get an experienced medical-device-channel commercial lawyer to draft or review the distributor agreement; the standard template that distributors offer is heavily distributor-favorable on these provisions.
Pattern four — classification dispute at submission. The foreign company self-classifies the device as Class II during diligence and submits the registration on that basis; NMPA's technical reviewers reclassify the device to Class III at intake, which restarts the project with the heavier clinical-trial and review requirements. The fix is the formal pre-classification consultation with NMPA's device-evaluation center during diligence, which provides a written classification determination before submission. The pre-classification consultation runs 2-4 months but eliminates the reclassification-at-intake risk.
The bundled engagement — sequence, indicative budget, realistic timeline
For a foreign medical-device or life-science company entering mainland China with a single device or product line, the bundled engagement covers these sequenced workstreams.
Phase one — regulatory diligence and pre-submission consultation (months 1-6). Device-classification confirmation through formal NMPA pre-classification consultation. Clinical-data gap analysis against current NMPA expectations. Quality-system gap analysis against NMPA inspection requirements. Mainland labeling and instructions-for-use adaptation. Mainland-specific post-market-surveillance plan drafting. Pre-submission strategy meeting with NMPA's relevant device-evaluation center.
Phase two — registration submission and review (months 4-30). Formal registration submission. Initial technical review. Q&A responses from NMPA reviewers. Mainland clinical trial if required for Class III. Quality-system inspection coordination. Final review and registration certificate issuance. This phase runs 12-30 months depending on device class and submission quality.
Phase three — commercial structure preparation (months 12-24, running parallel to phase two). Distributor selection process. Distributor agreement negotiation including the registration-ownership, transfer, and exit-rights provisions. Initial trademark filings in CNIPA for the device brand name. HK Ltd incorporation if applicable as the regional holding entity. CNIPA defensive filings for the brand name in relevant classes.
Phase four — commercial launch and validation (months 24-42). Distributor-channel commercial sales begin upon registration completion. Hospital-and-clinic channel relationships established. Post-market-surveillance reporting infrastructure operational. Mainland sales-data accumulation for market-demand validation. KOL (Key Opinion Leader) physician engagement programs activated where applicable.
Phase five — WFOE conversion (months 30-48 onwards, if direct-sales transition is chosen). WFOE registration mirroring the trading-WFOE pattern. NMPA registration transfer from distributor to WFOE. General-taxpayer status, customs registration, import-export operating right, SAFE FX registration. Sales-team build-out. Distributor relationship transition to sub-distributor or exit.
Indicative budget band: For a single-device mainland market entry of this profile, the first-five-years total budget commonly runs $300,000-2,500,000 all-in, with the wide range reflecting device-class-driven cost variation. NMPA registration agent fees and government charges: $50,000-300,000 depending on class. Mainland clinical trial if required: $200,000-1,500,000 depending on study size and complexity. Quality-system documentation and inspection-readiness work: $30,000-150,000. Distributor relationship setup and management: $20,000-80,000 in agent and legal fees. HK Ltd and CNIPA filings: $10,000-30,000. WFOE conversion if applicable: $40,000-150,000 plus ongoing operating overhead. These bands are indicative for a single device; multi-device portfolios scale accordingly.
Realistic timeline: 18-36 months to first commercial sale for Class II devices; 30-60 months for Class III devices. Most foreign medical-device founders underestimate this timeline by 6-12 months at project start. Plan with cash-flow models that reflect the regulatory wait, and confirm investor or board alignment on the multi-year horizon before committing the project. To model the budget against your specific device profile, run the expansion-budget estimator.
Case study match — the German manufacturer pattern adapted for regulated goods
While the chinaonramp case-study library does not yet include a medical-device-specific case study, the structural conversion pattern adapted for medical-device-regulated goods follows the German manufacturer Rep-Office-to-WFOE conversion closely. The German manufacturer case study covers a precision-parts trading WFOE — not regulated goods — but the parallel-track conversion sequencing, the EOR bridge for staff continuity, the general-taxpayer and customs-registration workstream, and the customer-side direct-invoicing dynamics all transfer to the medical-device WFOE conversion.
The medical-device-specific differences that overlay on top of the German-manufacturer pattern: the NMPA registration transfer is a multi-month workstream not present in the trading-WFOE case; the quality-system inspection readiness adds a parallel preparation track; the hospital-and-clinic channel relationships add a commercial-transition workstream that is more delicate than the industrial-customer transition; and the registered-capital sizing typically lands higher than for a trading WFOE to accommodate the inventory commitments that medical-device direct sales require. A more vertical-specific case study will land in a future content wave; in the meantime the industrial-manufacturer case study reads as the closest available playbook for the entity-structure portion of the engagement.
Working in this industry?
Tell us your constraints — we'll reply with the partner firm and filing sequence that fits this niche.
Frequently asked questions
Can foreign clinical-trial data be used for NMPA registration?
Increasingly yes for Class II and III devices, since the 2017 acceptance-of-foreign-data reforms. Class III still typically requires confirmatory China-based clinical trials, but the bar is lower than it used to be.
Distributor model or WFOE for direct sales?
Distributor for early-stage market validation (faster, no entity overhead). WFOE for direct sales once you have a clinical-evidence base and a sales team committed to the market. The transition usually happens 18-24 months into the engagement.
Why is this vertical's timeline so long?
NMPA registration alone is 12-24 months for Class II, 24-36 for Class III, with clinical-trial requirements compounding. The entity setup and sales-team build are short by comparison — regulatory is the binding path.
Do you have a relevant case study?
Yes — the case-studies index lists four anonymized engagements across DTC, SaaS, industrial, and creator personas.
Or skip the form
Book a call with Mike
30 minutes, Zoom or Tencent Meeting. No discovery-call gauntlet.